nf-core/eager
A fully reproducible and state-of-the-art ancient DNA analysis pipeline
22.10.6.
Learn more.
Introduction
nf-core/eager is a scalable and reproducible bioinformatics best-practise processing pipeline for genomic NGS sequencing data, with a focus on ancient DNA (aDNA) data. It is ideal for the (palaeo)genomic analysis of humans, animals, plants, microbes and even microbiomes.
Pipeline summary
- (Optionally) create reference genome indices for mapping (
bwa,samtools, andpicard) - Sequencing quality control (
FastQC,Falco) - Sequencing adapter removal, paired-end data merging (
AdapterRemoval) - Read mapping to reference using (
bwa aln,bwa mem,CircularMapper,bowtie2, ormapAD) - Post-mapping processing, statistics and conversion to bam (
samtools, andpreseq) - Ancient DNA C-to-T damage pattern visualisation (
DamageProfiler) - PCR duplicate removal (
DeDuporMarkDuplicates) - Post-mapping statistics and BAM quality control (
Qualimap) - Library Complexity Estimation (
preseq) - Overall pipeline statistics summaries (
MultiQC)
Additional Steps
Additional functionality contained by the pipeline currently includes:
Input
- Automatic merging of complex sequencing setups (e.g. multiple lanes, sequencing configurations, library types)
Preprocessing
- Illumina two-coloured sequencer poly-G tail removal (
fastp) - Post-AdapterRemoval trimming of FASTQ files prior mapping (
fastp) - Automatic conversion of unmapped reads to FASTQ (
samtools) - Host DNA (mapped reads) stripping from input FASTQ files (for sensitive samples)
aDNA Damage manipulation
- Damage removal/clipping for UDG+/UDG-half treatment protocols (
BamUtil) - Damaged reads extraction and assessment (
PMDTools) - Nuclear DNA contamination estimation of human samples (
angsd)
Genotyping
- Creation of VCF genotyping files (
GATK UnifiedGenotyper,GATK HaplotypeCallerandFreeBayes) - Creation of EIGENSTRAT genotyping files (
pileupCaller) - Creation of Genotype Likelihood files (
angsd) - Consensus sequence FASTA creation (
VCF2Genome) - SNP Table generation (
MultiVCFAnalyzer)
Biological Information
- Mitochondrial to Nuclear read ratio calculation (
MtNucRatioCalculator) - Statistical sex determination of human individuals (
Sex.DetERRmine)
Metagenomic Screening
- Low-sequenced complexity filtering (
BBdukorPRINSEQ++) - Taxonomic binner with alignment (
MALTorMetaPhlAn 4) - Taxonomic binner without alignment (
Kraken2,KrakenUniq) - aDNA characteristic screening of taxonomically binned data from MALT (
MaltExtract)
Functionality Overview
A graphical overview of suggested routes through the pipeline depending on context can be seen below.
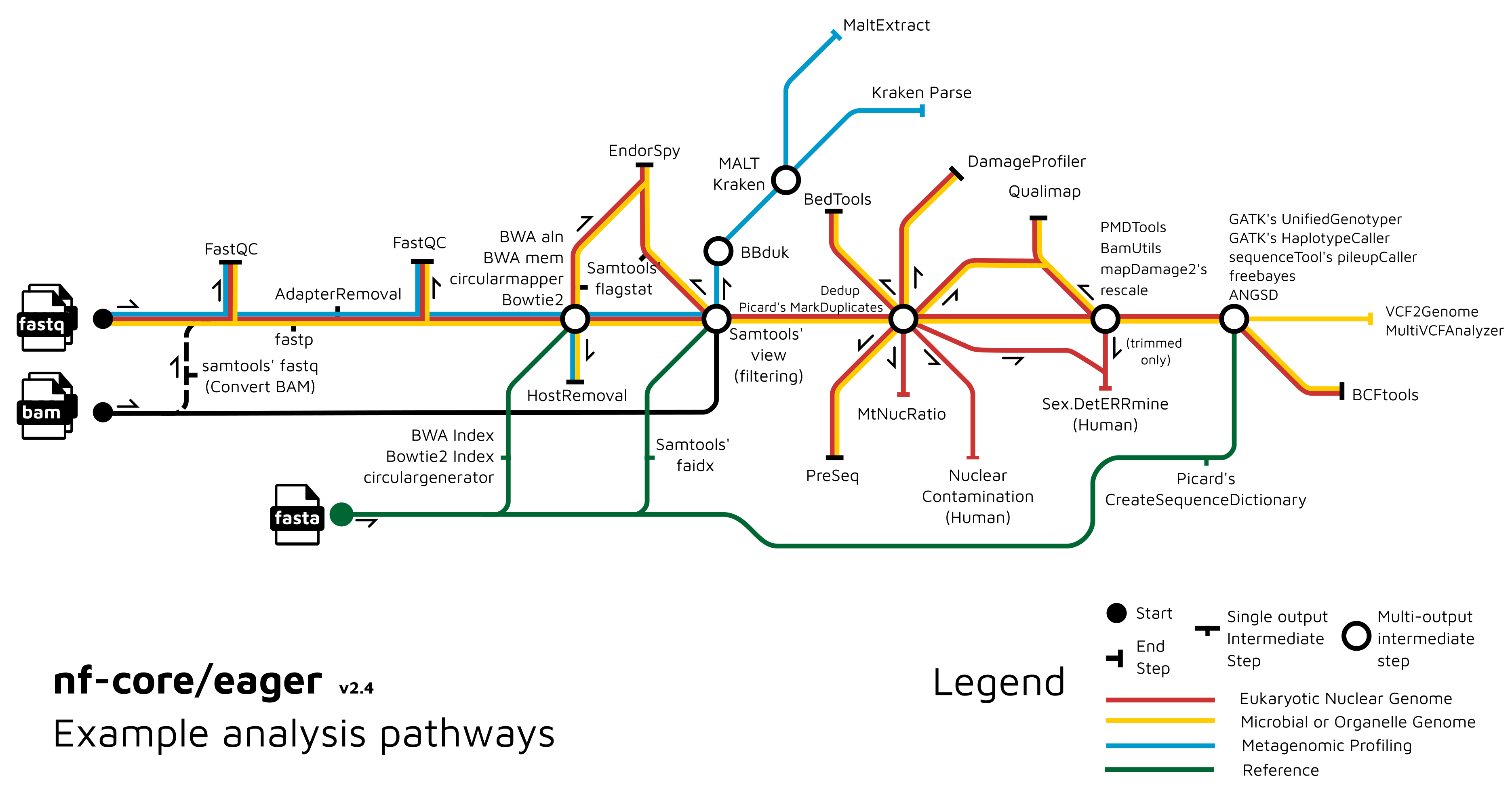
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.tsv:
ample_id library_id lane colour_chemistry pairment strandedness damage_treatment r1 r2 bam bam_reference_id
sample1 sample1_a 1 4 paired double none /<path>/<to>/sample1_a_l1_r1.fq.gz /<path>/<to>/sample1_a_l1_r2.fq.gz NA NA
sample2 sample2_a 2 2 single double full /<path>/<to>/sample2_a_l1_r1.fq.gz NA NA NA
sample3 sample3_a 8 4 single double half NA NA /<path>/<to>/sample31_a.bam Mammoth_MT_KrauseEach row represents a fastq file (single-end), pair of fastq files (paired end), and/or a bam file.
Now, you can run the pipeline using:
nextflow run nf-core/eager \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--fasta '<your_reference>.fasta' \
--outdir <OUTDIR>Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
This pipeline was established by Alexander Peltzer (apeltzer) and James A. Fellows Yates. Version two had major contributions from Stephen Clayton, Thiseas C. Lamnidis, Maxime Borry, Zandra Fagernäs, Aida Andrades Valtueña and Maxime Garcia and the nf-core community.
We thank the following people for their extensive assistance in the development of this pipeline:
- Alex Hübner
- Alexandre Gilardet
- Arielle Munters
- Åshild Vågene
- Charles Plessy
- Elina Salmela
- Fabian Lehmann
- He Yu
- Hester van Schalkwyk
- Ian Light-Máka
- Ido Bar
- Irina Velsko
- Işın Altınkaya
- Johan Nylander
- Jonas Niemann
- Katerine Eaton
- Kathrin Nägele
- Kevin Lord
- Luc Venturini
- Mahesh Binzer-Panchal
- Marcel Keller
- Megan Michel
- Merlin Szymanski
- Pierre Lindenbaum
- Pontus Skoglund
- Raphael Eisenhofer
- Roberta Davidson
- Rodrigo Barquera
- Selina Carlhoff
- Torsten Günter
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #eager channel (you can join with this invite).
Citations
If you use nf-core/eager for your analysis, please cite it using the following doi:
Fellows Yates JA, Lamnidis TC, Borry M, Valtueña Andrades A, Fagernäs Z, Clayton S, Garcia MU, Neukamm J, Peltzer A. 2021. Reproducible, portable, and efficient ancient genome reconstruction with nf-core/eager. PeerJ 9:e10947. DOI: 10.7717/peerj.10947.
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.